Tutorial 3: Slide-seq or Stereo-seq dataset¶
In this tutorial, we show how to apply scSTADE to identify spatial domains on Slide-seq or Stereo-seq. As a example, we analyse the Stereo dataset.
The source code package is freely available at https://github.com/cuiyaxuan/scSTADE/tree/master. The datasets used in this study can be found at https://drive.google.com/drive/folders/1H-ymfCqlDR1wpMRX-bCewAjG5nOrIF51?usp=sharing.
import sys
sys.path.append("/.../scSTADE-master/scSTADE_Cluster_Functions")
# Input the path.
from scSTADE import scSTADE
import os
import torch
import pandas as pd
import scanpy as sc
from sklearn import metrics
import multiprocessing as mp
def setup_seed(seed=41):
import torch
import os
import numpy as np
import random
torch.manual_seed(seed)
np.random.seed(seed) # Numpy module.
random.seed(seed) # Python random module.
if torch.cuda.is_available():
# torch.backends.cudnn.benchmark = False
torch.backends.cudnn.deterministic = True
torch.cuda.manual_seed(seed)
torch.cuda.manual_seed_all(seed)
#os.environ['PYTHONHASHSEED'] = str(seed)
setup_seed(41)
device = torch.device('cuda:0' if torch.cuda.is_available() else 'cpu')
n_clusters = 10 ###### the number of spatial domains.
file_path = '/home/cuiyaxuan/spatialLIBD/5.Mouse_Olfactory/' #please replace 'file_path' with the download path
adata = sc.read_h5ad(file_path + 'filtered_feature_bc_matrix.h5ad') #### project name
adata.var_names_make_unique()
model = scSTADE(adata,datatype='Slide',device=device,n_top_genes=4000)
adata = model.train()
radius = 50
tool = 'mclust' # mclust, leiden, and louvain
from utils import clustering
if tool == 'mclust':
clustering(adata, n_clusters, radius=radius, method=tool, refinement=True)
elif tool in ['leiden', 'louvain']:
clustering(adata, n_clusters, radius=radius, method=tool, start=0.1, end=2.0, increment=0.01, refinement=False)
adata.obs['domain']
adata.obs['domain'].to_csv("label.csv")
/home/cuiyaxuan/anaconda3/envs/pytorch/lib/python3.8/site-packages/scanpy/preprocessing/_highly_variable_genes.py:62: UserWarning: flavor='seurat_v3' expects raw count data, but non-integers were found. warnings.warn(
Graph constructed!
Building sparse matrix ...
Begin to train ST data...
0%| | 0/500 [00:00<?, ?it/s]
0
0%| | 1/500 [00:00<04:24, 1.89it/s]
0
0%|▏ | 2/500 [00:00<04:01, 2.07it/s]
0
1%|▎ | 3/500 [00:01<03:53, 2.13it/s]
0
1%|▎ | 4/500 [00:01<03:49, 2.16it/s]
100%|█████████████████████████████████████████| 500/500 [03:49<00:00, 2.17it/s]
Optimization finished for ST data!
R[write to console]: __ __ ____ ___ _____/ /_ _______/ /_ / __ `__ / ___/ / / / / ___/ __/ / / / / / / /__/ / /_/ (__ ) /_ /_/ /_/ /_/___/_/__,_/____/__/ version 6.0.0 Type 'citation("mclust")' for citing this R package in publications.
fitting ...
|======================================================================| 100%
import matplotlib as mpl
import scanpy as sc
import numpy as np
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
import warnings
import visual_high
mpl.rcParams['pdf.fonttype'] = 42
mpl.rcParams["font.sans-serif"] = "Arial"
warnings.filterwarnings('ignore')
file_path = '/home/cuiyaxuan/spatialLIBD/5.Mouse_Olfactory/' #please replace 'file_path' with the download path
adata = sc.read_h5ad(file_path + 'filtered_feature_bc_matrix.h5ad') #### project name
df_label=pd.read_csv('./label.csv', index_col=0)
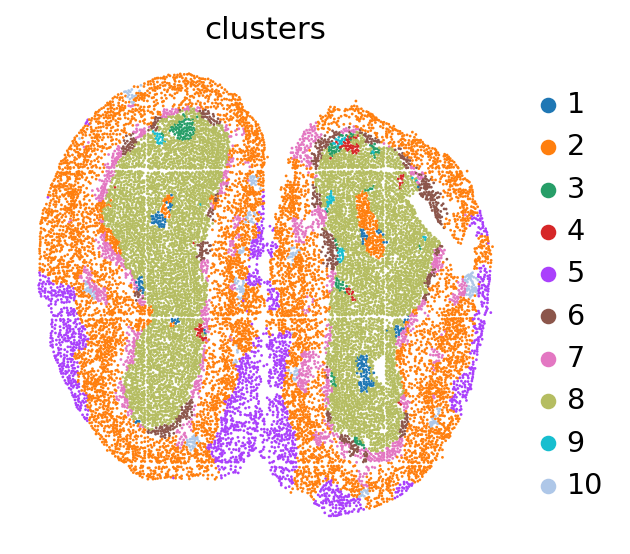
visual_high.visual(adata,df_label)
#cells after MT filter: 19109
WARNING: saving figure to file figures/spatialHippocampus.pdf