Tutorial 5: Identifying FVGs in 10X Visium DLPFC dataset¶
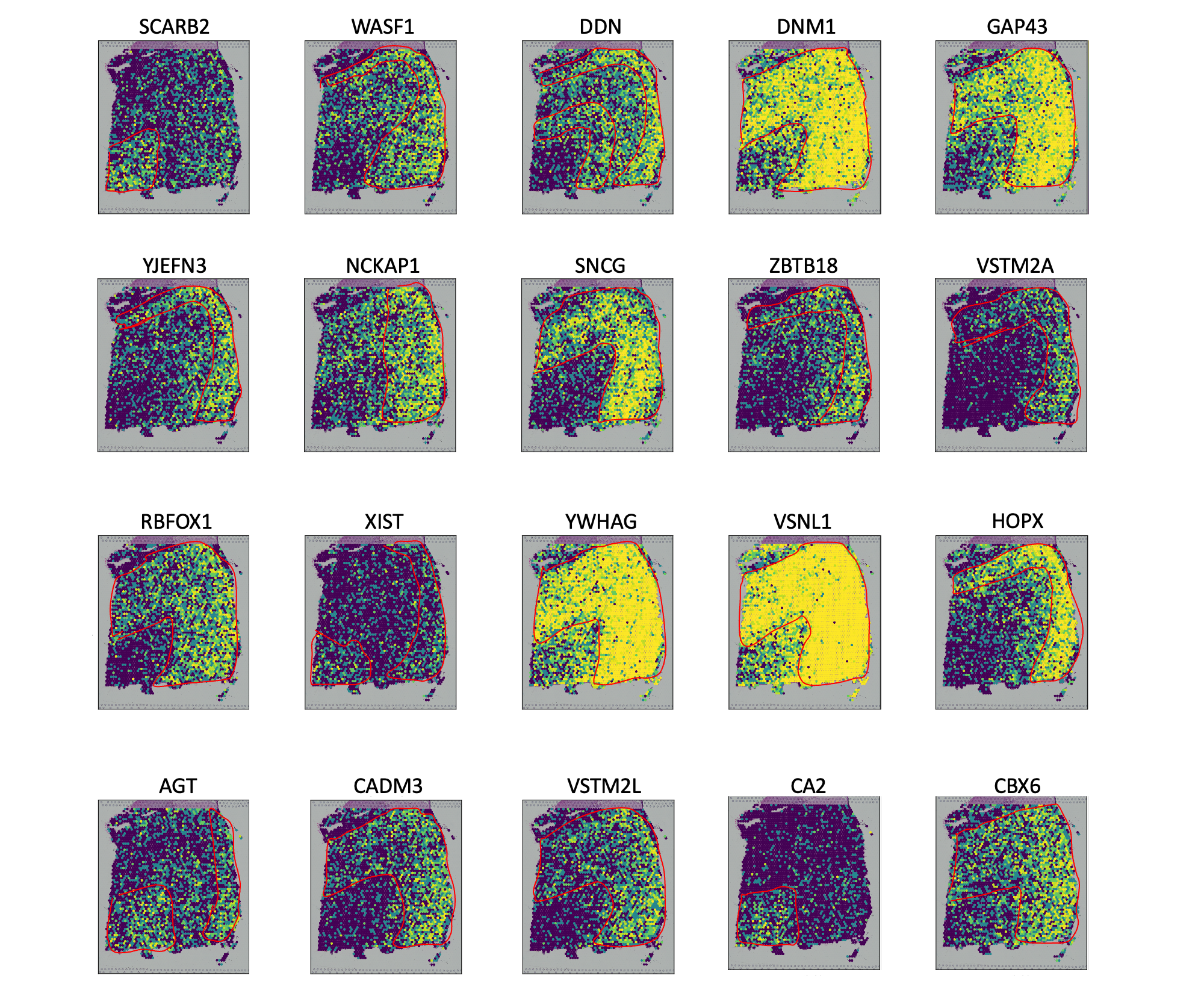
In this tutorial, we show how to apply scSTADE to identify FVGs on 10X Visium DLPFC dataset. As a example, we analyse the Visium dataset.
The source code package is freely available at https://github.com/cuiyaxuan/scSTADE/tree/master. The datasets used in this study can be found at https://drive.google.com/drive/folders/1H-ymfCqlDR1wpMRX-bCewAjG5nOrIF51?usp=sharing.
First, cd /home/.../scSTADE_FVG_Functions/div>
# we execute the FVG model in the R environment.
conda create -n r4
source activate r4
conda search r-base
conda install r-base=4.2.0
conda install conda-forge::r-seurat==4.4.0
conda install conda-forge::r-hdf5r
onda install r-devtools
Using R virtual environment with conda。
library("devtools")
devtools::install_github("shaoqiangzhang/DEGman")
install.packages("hdf5r")
install.packages('philentropy')
install.packages('dplyr')
install.packages('foreach')
install.packages('parallel')
install.packages('doParallel')
# You can find the estimate clustering labels and the running results in the scSTADE/scSTADE_FVG_Functions/.
# You can run "Rscript step1_run_DEG.R"
# =========================
# 1. Load libraries
# =========================
source("functions_DEG.R")
library(DEGman)
library(Seurat)
library(dplyr)
library(hdf5r)
library(philentropy)
library(foreach)
library(doParallel)
library(doSNOW)
# =========================
# 3. Load data
# =========================
hc1 <- Read10X_h5(
"/home/cuiyaxuan/spatialLIBD/151673/151673_filtered_feature_bc_matrix.h5" #### to your path and project name
)
label <- read.csv(
"/home/cuiyaxuan/metric_change/revise_R2/est_151673/conlabel.csv", #### to your path of cluster label
header = TRUE,
row.names = 1
)
# =========================
# 4. Preprocess (once)
# =========================
matlist <- prep_matlist(hc1, label)
k <- length(matlist) # number of clusters
# =========================
# 5. Parallel + progress bar
# =========================
ncore <- min(k, parallel::detectCores() - 1)
cl <- makeCluster(ncore)
registerDoSNOW(cl)
pb <- txtProgressBar(min = 0, max = k, style = 3)
progress <- function(n) setTxtProgressBar(pb, n)
opts <- list(progress = progress)
files <- foreach(
n = 1:k,
.packages = c("DEGman"),
.options.snow = opts
) %dopar% {
run_one_cluster(matlist, n)
}
close(pb)
stopCluster(cl)
# =========================
# 6. Done
# =========================
#################################Functionally variable genes weight compute################################
# You can run "Rscript step2_run_FVG.R"
source("functions_FVG.R")
library(tidyverse)
rounded_number <- 3
fre <- list.files(
path = ".",
pattern = "\\.csv$",
full.names = TRUE
) %>%
map_dfr(read.csv) %>% # 自动 rbind
count(x) %>% # table(vec1$x)
filter(n >= rounded_number) %>%
pull(x)
write.csv(fre, "df1.csv", row.names = FALSE)
# ==============================================================================
# File: main.R
# Description: Main execution script for FVG calculation
# Usage: Make sure functions_FVG.R is in the same directory before running
# ==============================================================================
library(Seurat)
library(dplyr)
library(hdf5r)
library(foreach)
library(parallel)
library(doParallel)
# ==============================================================================
# 1. Set paths and load data
# ==============================================================================
h5_path <- "/home/cuiyaxuan/spatialLIBD/151673/151673_filtered_feature_bc_matrix.h5" #### to your path and project name
spatial_path <- "/home/cuiyaxuan/spatialLIBD/151673/spatial/tissue_positions_list.csv" #### to your path and project name
gene_list_path <- "df1.csv"
cat("Loading data...\n")
hc1 <- Read10X_h5(h5_path)
tissue_local <- read.csv(spatial_path, row.names = 1, header = FALSE)
# ==============================================================================
# 2. Seurat preprocessing
# ==============================================================================
pbmc <- CreateSeuratObject(counts = hc1, project = "HC_1", min.cells = 10)
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 30000)
# ==============================================================================
# 3. Data alignment
# ==============================================================================
common_cells <- intersect(colnames(pbmc), rownames(tissue_local))
cat(sprintf("Common cells found: %d\n", length(common_cells)))
pbmc <- subset(pbmc, cells = common_cells)
tissue_local <- tissue_local[common_cells, ]
# Load target gene list
df1 <- read.csv(gene_list_path, header = TRUE)
target_genes <- df1[, 1]
# Keep genes present in expression matrix
valid_genes <- intersect(target_genes, rownames(pbmc))
# Extract expression matrix (cells × genes)
mat_sparse <- GetAssayData(pbmc, layer = "data")[valid_genes, ]
dat1 <- as.matrix(t(mat_sparse))
# Extract spatial coordinates (first two columns)
x_y_list <- tissue_local[, 1:2]
if (!all(rownames(dat1) == rownames(x_y_list))) {
stop("Data alignment error!")
}
# ==============================================================================
# 4. Parallel computation
# ==============================================================================
n_cores <- 24
cat(sprintf("Starting parallel processing with %d cores...\n", n_cores))
cls <- makeCluster(n_cores)
registerDoParallel(cls)
# Export required objects to worker nodes
clusterExport(cls, c("cripar_opt", "getmode", "x_y_list"))
crinum <- foreach(
gene_vec = iter(dat1, by = "col"),
.combine = "c",
.packages = "stats"
) %dopar% {
cripar_opt(gene_vec, x_y_list, Ccri = 10, highval = 500, lowval = 50) # Users can freely adjust the following three parameters: Ccri = 10, highval = 500, and lowval = 50.
}
stopCluster(cls)
# ==============================================================================
# 5. Save results
# ==============================================================================
result_df <- data.frame(gene = valid_genes, crinum = crinum)
# Merge with original gene list (keep original order)
final_res <- merge(df1, result_df,
by.x = names(df1)[1],
by.y = "gene",
all.x = TRUE)
fvg_sort <- final_res[order(-final_res$crinum), ]
write.csv(fvg_sort, "fvg.csv", row.names = FALSE)
cat("Done! Results saved to 'fvg.csv'.\n")
/home/cuiyaxuan/anaconda3/envs/pytorch/lib/python3.8/site-packages/tqdm/auto.py:22: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
/home/cuiyaxuan/anaconda3/envs/pytorch/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call .var_names_make_unique.
utils.warn_names_duplicates("var")
/home/cuiyaxuan/anaconda3/envs/pytorch/lib/python3.8/site-packages/scanpy/readwrite.py:413: DtypeWarning: Columns (1,2,3,4,5) have mixed types. Specify dtype option on import or set low_memory=False.
positions = pd.read_csv(files['tissue_positions_file'], header=None)